甲状腺眼症治療用IGF-1R阻害剤テッペーザの有効性と安全性
活動性甲状腺眼症(TED)患者を対象とした海外第Ⅲ相試験(海外データ)
承認時評価資料:活動性甲状腺眼症患者を対象とした海外第Ⅲ相試験(HZNP-TEP-301試験)(2024年9月24日承認、CTD 2.7.6.4)
Douglas R.S. et al.: N Engl J Med 2020; 382:341-352.
COI:本試験はAMGEN社(旧HorizonTherapeutics社)の資金により行われた。
著者にAMGEN社(旧HorizonTherapeutics社)よりコンサルタント料等を受領した者、同社の株式を保有している者及び社員が含まれる。
(1)試験概要
目的
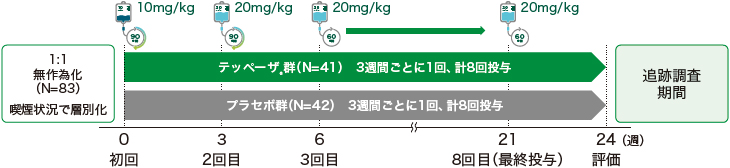
中等症から重症の活動性甲状腺眼症(TED)患者にテッペーザ®を3週間間隔で24週間投与したときの有効性及び安全性をプラセボ投与時と比較、検討する。
試験デザイン
第Ⅲ相、無作為化、二重遮蔽、プラセボ対照、並行群間比較、多施設共同試験
対象
中等症から重症の活動性甲状腺眼症患者83例(テッペーザ®群41例、プラセボ群42例)
主な選択基準は以下のとおりとした。
- 試験眼のCASが4点以上
- 活動性甲状腺眼症の発症後9ヵ月未満
- 甲状腺機能が正常な患者、又は軽度の甲状腺機能低下症もしくは甲状腺機能亢進症を有する患者(FT4及びFT3の正常値からの逸脱が50%未満)
等
なお、過去に甲状腺眼症の治療を目的とした眼窩への放射線療法又は外科的療法を受けた患者は除外した。副腎皮質ステロイド(最大累積用量がメチルプレドニゾロン又はその同等薬で1g未満)による治療歴を有する患者は、スクリーニングの4週間以上前に副腎皮質ステロイド投与を中止していれば組入れ可能とした。
試験方法
- 本試験は、スクリーニング期間(4週間)、投与期間(24週間)、及び追跡調査期間(48週間)から構成された。
- 対象患者をDay 1に喫煙状況(喫煙者、非喫煙者)により層別化して、テッペーザ®又はプラセボに1:1の比で無作為に割り付け、二重遮蔽投与期間に3週間間隔で8回投与した。
- テッペーザ®の用量を、初回投与では10mg/kg、その後7回の投与では20mg/kgとした。
- 初回及び2回目投与では約90分かけて投与し、注入に伴う事象が見られなかった場合は、3回目以降の投与では約60分かけて投与した。
- 二重遮蔽投与期間終了時(投与24週)の眼球突出非奏効例(試験眼で眼球突出の減少が2mm未満)はオープンラベル継続試験[HZNP-TEP-302試験(OPTIC-X試験)]へ移行した。
- 眼球突出奏効例及びOPTIC-X試験への参加を選択しなかった眼球突出非奏効例は追跡調査期間へ移行した。
- 投与24週時の眼球突出奏効例のうち追跡調査期間中に再発の基準を満たした患者を、OPTIC-X試験に組入れ可能とした。再発の基準を、投与24週以降に試験眼で眼球突出が投与24週時から2mm以上増加、又はCASが4点以上かつ投与24週時から2点以上増加した場合とした。本基準に加えて、治験責任医師が患者の症状から再発を確認した(新たな複視の発現など)。
- 追跡調査期間の最終来院(投与72週)を完了した患者を対象に、6ヵ月後及び12ヵ月後に最終来院以降の甲状腺眼症治療の有無を治験実施スタッフが電話又はメールで確認した(追跡調査連絡期間)。
評価項目
ベースライン時(Day 1)に重症度が高いと判断された側の眼を「試験眼」とし、逆側の眼を「僚眼」とした。両眼の重症度が同程度の場合、治験責任医師が試験眼を選択した。両眼で有効性を評価したが、主要解析には試験眼を使用した。
[主要評価項目]
- 投与24週時の眼球突出奏効率[試験眼の眼球突出がベースラインから2mm以上減少し、かつ僚眼の眼球突出の悪化(2mm以上の増加)が認められない患者の割合]
[副次評価項目]
- 投与24週時の全般的奏効率[ベースラインと比べて、試験眼で眼球突出が2mm以上減少かつCASが2点以上減少し、さらに僚眼で眼球突出又はCASの悪化(眼球突出の2mm以上の増加又はCASの2点以上の増加)が認められない患者の割合]
- 投与24週時のCAS奏効率[試験眼のCASが0点又は1点(炎症所見なし又は軽微な炎症所見)であった患者の割合]
- 投与24週時までの試験眼での眼球突出のベースラインからの平均変化量
- 投与24週時の複視奏効率[ベースラインに試験眼で複視がグレード1以上であった患者のうち、試験眼で1グレード以上減少し、かつ僚眼で悪化(1グレード以上の増加)が認められなかった患者の割合]
- 投与24週時までのGO-QoL(総合スコア)のベースラインからの平均変化量
[安全性評価項目]
- 有害事象、免疫原性 等
解析計画
以下の解析対象集団を定義した。
-
ITT(intent-to-treat)集団:
治験薬(テッペーザ®又はプラセボ)に無作為に割り付けられた全ての患者。全ての有効性評価項目について、ITT集団を用いて割り付けた投与に基づき解析した。 -
安全性解析対象集団:
治験薬(テッペーザ®又はプラセボ)を1回以上投与された全ての患者。安全性評価項目について、安全性解析対象集団を用いて投与された治験薬に基づき解析した。
主要評価項目及び副次評価項目を階層的手順で逐次検定し、テッペーザ®群とプラセボ群を比較した(有意水準0.05)。プラセボ群と比べてテッペーザ®群で投与群間に統計学的有意差が示された場合にのみ、次の評価項目を検定した。
なお、主要評価項目、副次評価項目において、追加解析として投与群及び来院別(投与6、12、18
週時)に各項目の解析を実施した。
主要評価項目
- 眼球突出奏効率の主要解析では、無作為化に用いた層別因子である喫煙状況(喫煙者、非喫煙者)で層別して眼球突出奏効率の群間差を算出し、各層の推定量をCochran-Mantel-Haenszel(CMH)の重みを用いて併合して、標準誤差で除すことによって検定統計量を算出した。また、検定統計量が標準正規分布であると仮定して帰無仮説のもとで両側p値を算出した。
- 投与24週時の評価が欠測であった患者は主要解析で非奏効例とみなした。また、投与期間の投与21週より前に治験薬投与を中止した患者は、投与24週時の評価結果が得られた場合を除き、非奏効例とみなした。
[副次評価項目]
- 全般的奏効率、CAS奏効率、及び複視奏効率の主要解析には主要評価項目と同じ方法を用いた。
- 眼球突出及びGO-QoL(総合スコア)の変化量は、試験眼でのベースラインからの変化量(個別値)に適合する共分散分析モデルの反復測定混合モデルを用いて解析した。モデルには、ベースライン値、喫煙状況(喫煙者、非喫煙者)、投与群、評価時点、評価時点と投与群の交互作用、及び評価時点とベースライン値の交互作用を含め、構造化されていない分散共分散行列を用いた。
-
副次評価項目の検定順序は以下とした。
1)全般的奏効率、2)CAS奏効率、3)眼球突出のベースラインからの変化量、4)複視奏効率、5)GO-QoL(総合スコア)のベースラインからの変化量
投与24週時の主要評価項目及び副次評価項目の結果
[安全性評価項目]
- 安全性の解析では記述統計量のみを算出した。
- 有害事象をICH国際医薬用語集(MedDRA)Version 20.1を用いてコード化した。
- 治験薬初回投与(投与1日)から投与期間の終了[最終投与3週(21日)後]までに発現した事象又は悪化した事象を治験薬投与下で発現した有害事象(TEAE)と定義した。
- TEAE及び追跡調査有害事象を器官別大分類(SOC)及び基本語(PT)別に投与群ごとに示した。各TEAEと治験薬との因果関係を、治験責任医師及び治験依頼者が分類した。注目すべきTEAEとして潜在的な注入に伴う反応、アナフィラキシー反応、聴覚障害、高血糖、筋痙縮、及び下痢を解析し、注目すべき追跡調査有害事象として聴覚障害、高血糖、筋痙縮、及び下痢を解析した。
- テッペーザ®群で抗薬物抗体(ADA)陽性であった患者の割合を示した。ADA陽性が確認された検体を1つ以上有した患者をADA陽性例とした。
CAS:Clinical Activity Score
GO-QoL:バセドウ病眼症の生活の質に関する質問票
(2)患者背景
■人口統計学的特性及びベースラインの疾患特性(安全性解析対象集団)
| テッペーザ®群 (N=41t) |
プラセボ群 (N=42) |
||
|---|---|---|---|
| 年齢(歳)、平均値±標準偏差 | 51.6±12.63 | 48.9±12.96 | |
| 年齢区分、 n(%) |
65歳未満 | 32(78.0) | 38(90.5) |
| 65歳以上 | 9(22.0) | 4(9.5) | |
| 性別、 n(%) |
女性 | 29(70.7) | 31(73.8) |
| 男性 | 12(29.3) | 11(26.2) | |
| 人種、 n(%) |
アジア人 | 2(4.9) | 1(2.4) |
| 黒人/アフリカ系アメリカ人 | 4(9.8) | 2(4.8) | |
| 白人 | 35(85.4) | 37(88.1) | |
| その他 | 0 | 2(4.8) | |
| 身長(cm)、平均値±標準偏差 | 167.7±10.21 | 167.4±10.22 | |
| 体重(kg)、平均値±標準偏差 | 75.0±16.54 | 75.8±18.51 | |
| BMI(kg/m2)、平均値±標準偏差 | 26.7±5.63 | 26.9±5.30 | |
| 喫煙歴 n(%) |
過去に喫煙歴あり | 9(22.0) | 9(21.4) |
| 喫煙者 | 9(22.0) | 8(19.0) | |
| 喫煙歴なし | 23(56.1) | 25(59.5) | |
| 飲酒歴 n(%) |
過去に喫煙歴あり | 3(7.3) | 3(7.1) |
| 飲酒習慣あり | 21(51.2) | 24(57.1) | |
| 飲酒習慣なし | 17(41.5) | 15(35.7) | |
| HbA1c(%)、平均値±標準偏差 | 5.5±0.42 | 5.5±0.39 | |
| バセドウ病の罹病期間(年)、中央値[範囲] | 1.0[0.26-28.24] | 0.9[0.09-14.81] | |
| 甲状腺眼症の罹病期間(月)、中央値[範囲] | 6.3[0.92-9.67] | 6.8[1.05-10.33] | |
(3)有効性

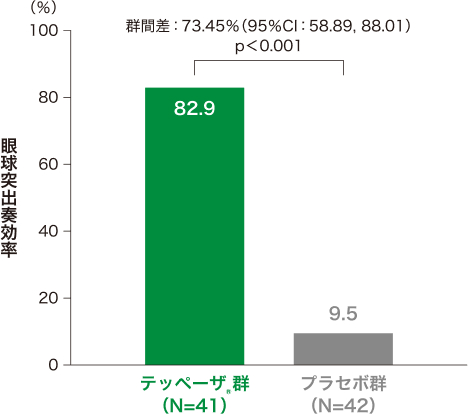
1)投与24週時の眼球突出奏効率*[主要評価項目、検証的解析結果]
投与24週時の眼球突出奏効率はテッペーザ®群で82.9%、プラセボ群で9.5%であり、テッペーザ®群で統計学的に有意な改善が認められ(p<0.001、検定統計量が標準正規分布であると仮定して両側p値を算出)、優越性が検証されました。
■投与24週時の眼球突出奏効率*(試験眼、ITT集団)
■投与24週時及び来院別の眼球突出奏効率*(試験眼、ITT集団)※投与6週時から18週時は名目上のp値
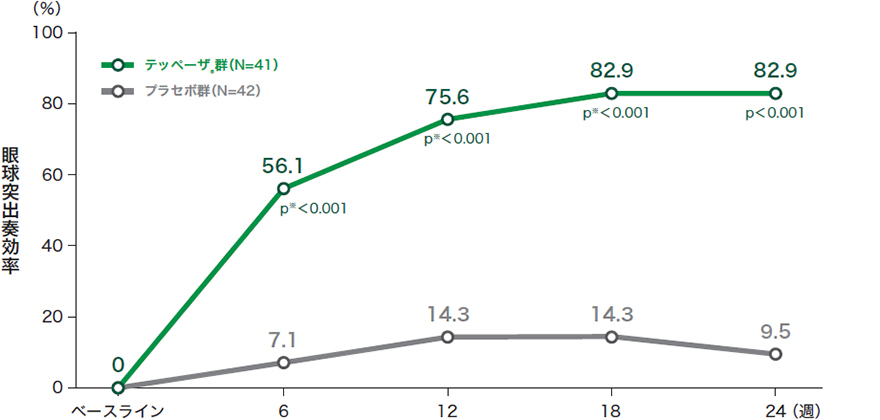
投与24週時のp値:プラセボとの比較 喫煙状況で層別化したCMH検定を用いて算出、有意水準両側5%
ITT:intent-to-treat(いずれかの治療群に割り付けられたすべての患者)
以下、事前規定されていない評価項目ですが、承認時に評価された結果のため掲載いたします。
■眼球突出奏効率の喫煙状況別(試験眼、ITT集団)
眼球突出奏効率は非喫煙者(テッペーザ®群84.4%、プラセボ群8.8%、p<0.001)及び喫煙者(テッペーザ®群77.8%、プラセボ群12.5%、p=0.007)のいずれもプラセボ群と比べてテッペーザ®群で高くなりました。
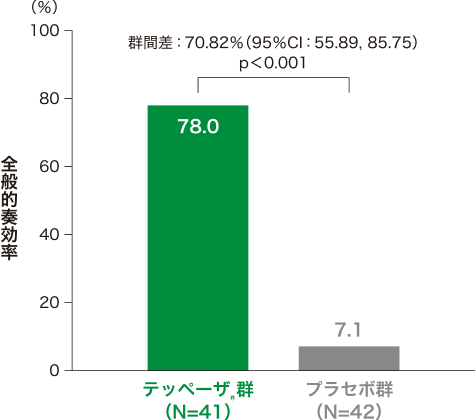
2)投与24週時の全般的奏効率*[副次評価項目]
投与24週時の全般的奏効率はテッペーザ®群で78.0%、プラセボ群で7.1%であり、統計学的な有意差が認められました(p
<0.001、検定統計量が標準正規分布であると仮定して両側p値を算出)。
■投与24週時の全般的奏効率*(試験眼、ITT集団)
■投与24週時及び来院別の全般的奏効率*(試験眼、ITT集団)※投与6週時から18週時は名目上のp値
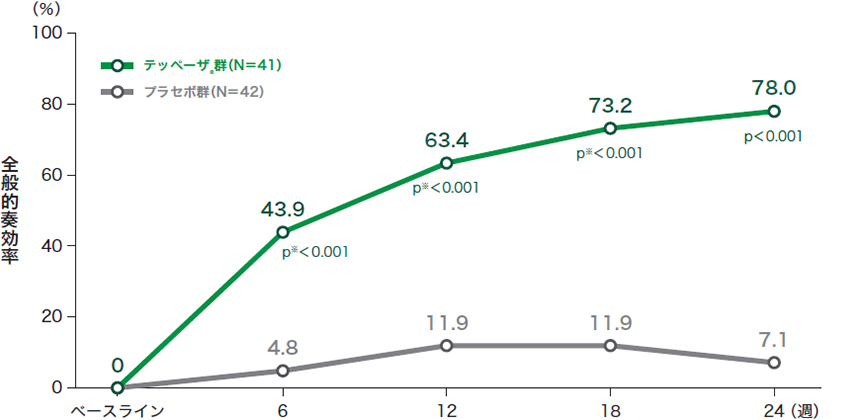
投与24週時のp値:プラセボとの比較 喫煙状況で層別化したCMH検定を用いて算出、有意水準両側5%
ITT:intent-to-treat(いずれかの治療群に割り付けられたすべての患者)
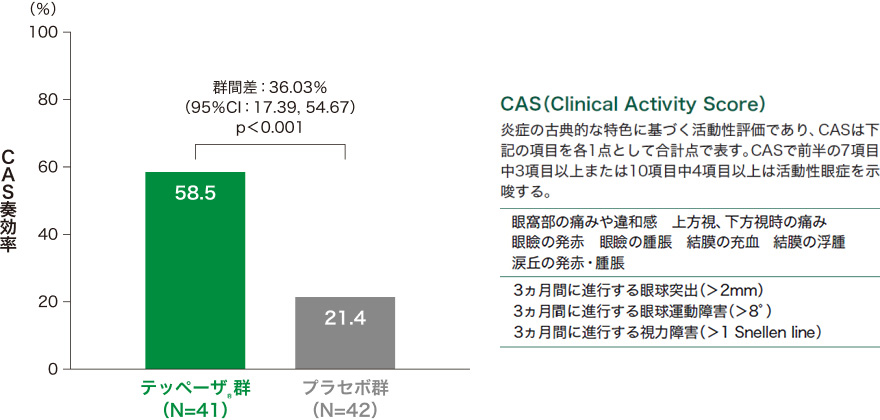
3)投与24週時のCAS奏効率*[副次評価項目]
投与24週時のCAS奏効率はテッペーザ®群で58.5%、プラセボ群で21.4%であり、統計学的な有意差が認められました
(p<0.001、検定統計量が標準正規分布であると仮定して両側p値を算出)。
■投与24週時のCAS奏効率*(試験眼、ITT集団)
■投与24週時及び来院別のCAS奏効率*(試験眼、ITT集団)※投与6週時から18週時は名目上のp値
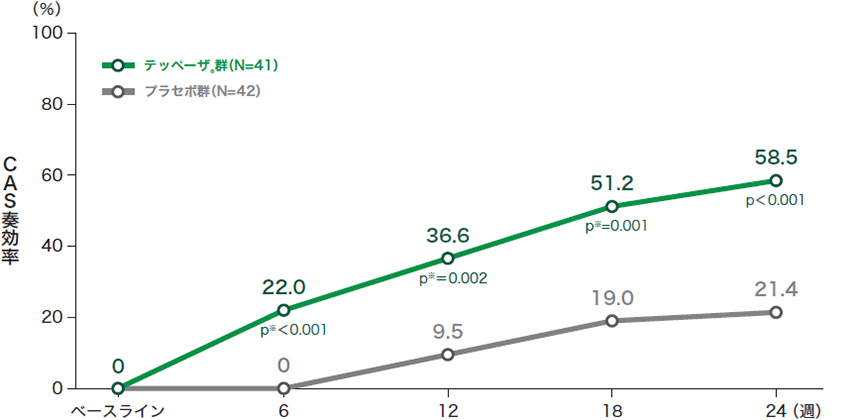
*試験眼のCASが0点又は1点(炎症所見なし又は軽微な炎症所見)であった患者の割合
ITT:intent-to-treat(いずれかの治療群に割り付けられたすべての患者) CAS:Clinical Activity Score
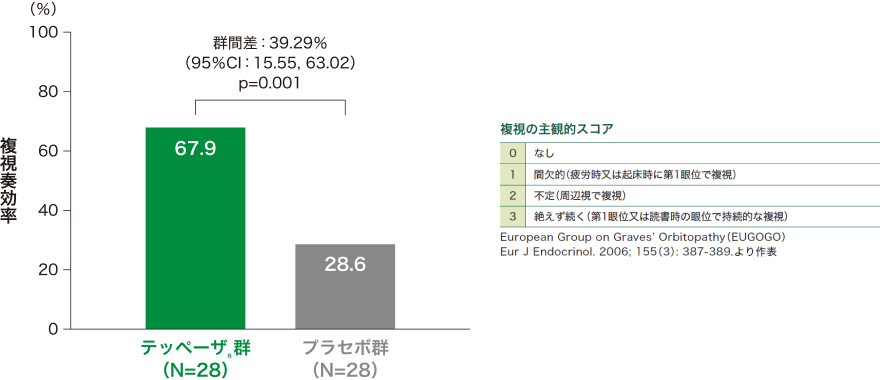
4)投与24週時の複視奏効率*[副次評価項目]
投与24週時の複視奏効率はテッペーザ®群で67.9%、プラセボ群で28.6%であり、統計学的な有意差が認められました
(p=0.001、検定統計量が標準正規分布であると仮定して両側p値を算出)。
■投与24週時の複視奏効率*(ITT集団のうちベースラインに複視が認められた患者)
■投与24週時及び来院別の複視奏効率*(試験眼、ITT集団のうちベースラインに複視が認められた患者)
※投与6週時から18週時は名目上のp値
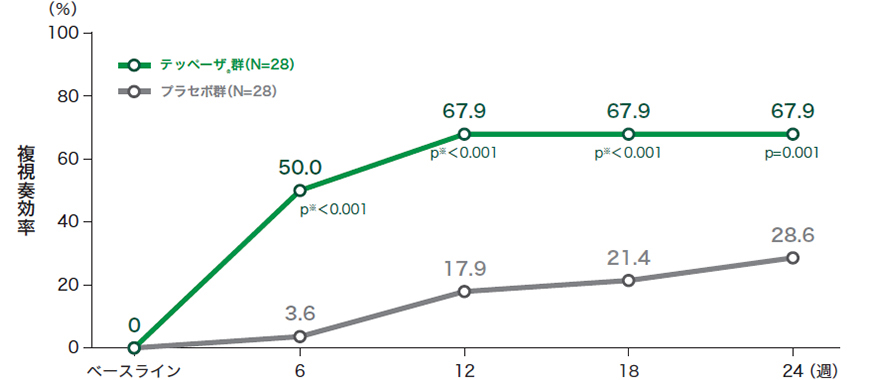
た患者の割合
投与24週時のp値:プラセボとの比較 喫煙状況で層別化したCMH検定を用いて算出、有意水準両側5%
ITT:intent-to-treat(いずれかの治療群に割り付けられたすべての患者)
追跡調査期間における各評価
眼球突出奏効率
追跡調査期間におけるテッペーザ®群の眼球突出奏効率は84.8%~90.9%であり、投与72週時では90.5%(19/21例)でした。
テッペーザ®群の眼球突出奏効持続率※は投与28週時で91.2%(31/34例)、投与72週時で55.9%(19/34例)でした。
全般的奏効率
追跡調査期間におけるテッペーザ®群の全般的奏効持続率※は投与28週時で90.6%(29/32例)、投与72週時で56.3%
(18/32例)でした。
CAS奏効率
追跡調査期間におけるテッペーザ®群のCAS奏効率は56.0%~72.7%であり、投与72週時では66.7%(14/21例)でした。テッペーザ®群のCAS奏効持続率※は、投与28週時で91.7%(22/24例)、投与72週時で50.0%(12/24例)でした。
※ 投与24週時のCAS奏効例に対して、追跡調査期間の各来院時に奏効が持続しており、かつ他の甲状腺眼症治療を受けていなかった患者の割合を「CAS奏効持続率」と定義した。眼球突出のベースラインからの平均変化量
追跡調査期間における投与72週時までのテッペーザ®群(21例)の眼球突出のベースラインからの平均変化量は―3.62mmでした。
複視奏効率
追跡調査期間におけるテッペーザ®群の複視奏効率は62.5%~80.0%であり、投与72週時では、80.0%(12/15例)でした。 テッペーザ®群の複視奏効持続率※は投与28週時で73.7%(14/19例)、投与72週時で57.9%(11/19例)でした。
※ 投与24週時の複視奏効例に対して、追跡調査期間の各来院時に奏効が持続しており、かつ他の甲状腺眼症治療を受けていなかった患者の割合を「複視奏効持続率」と定義した。GO-QoL(総合スコア)のベースラインからの変化量
追跡調査期間では、投与72週時のテッペーザ®群(21例)のGO-QoL(総合スコア)のベースラインからの変化量は21.19でした。
(4)安全性
二重遮蔽投与期間
- 副作用は、テッペーザ®群で63.4%(26/41例)、プラセボ群で26.2%(11/42例)に認められました。
- 主な副作用は、テッペーザ®群では筋痙縮29.3%(12/41例)、脱毛症24.4%(10/41例)、下痢、悪心、口内炎、頭痛、無月経、皮膚乾燥、及び睫毛眉毛脱落症が各7.3%(3/41例)でした。
- 重大な副作用は、テッペーザ®群で注入に伴う反応が1例に認められ、投与中止に至りました。
- 本試験の二重遮蔽期間に死亡は認められませんでした。
■二重遮蔽投与期間:いずれかの投与群で2例以上に発現した副作用(安全性解析対象集団)
|
テッペーザ®群 (N=41) |
プラセボ群 (N=42) |
|
|---|---|---|
| 副作用 | 26(63.4) | 11(26.2) |
| 耳および迷路障害 | 4(9.8) | 0 |
| 耳不快感 | 2(4.9) | 0 |
| 胃腸障害 | 9(22.0) | 7(16.7) |
| 上腹部痛 | 0 | 2(4.8) |
| 下痢 | 3(7.3) | 4(9.5) |
| 悪心 | 3(7.3) | 3(7.1) |
| 口内炎 | 3(7.3) | 1(2.4) |
| 筋骨格系および 結合組織障害 |
12(29.3) | 3(7.1) |
| 筋痙縮 | 12(29.3) | 2(4.8) |
|
MedDRA Version.20.1 |
|
テッペーザ®群 (N=41) |
プラセボ群 (N=42) |
|
|---|---|---|
| 神経系障害 | 7(17.1) | 1(2.4) |
| 浮動性めまい | 2(4.9) | 0 |
| 頭痛 | 3(7.3) | 1(2.4) |
| 生殖系および乳房障害 | 3(7.3) | 0 |
| 無月経 | 3(7.3) | 0 |
| 皮膚および 皮下組織障害 |
15(36.6) | 4(9.5) |
| 脱毛症 | 10(24.4) | 3(7.1) |
| 皮膚乾燥 | 3(7.3) | 0 |
| 毛髪成長異常 | 2(4.9) | 0 |
| 睫毛眉毛脱落症 | 3(7.3) | 0 |
|
n(%) |
追跡調査期間
- 副作用の発現割合は、テッペーザ群®で19.4%(7/36例)、プラセボ群は0%(0/4例)でした。
- 二重遮蔽投与期間の追跡調査期間に重篤な副作用及び死亡は認められませんでした。